Chediak-Higashi syndrome
Chediak-Higashi syndrome
Updated: 08/03/2020
© Jun Wang,
MD, PhD
General features
- Rare
- Autosomal recessive
- Lysosomal storage disorder
- Associated with primary immunodeficiency due to impaired phagocytosis
- Affects multiple systems
- Symptoms usually present soon after birth
- Most patients died before 10 as a result of infection or an accelerated lymphoma like phase
Pathogenesis
- Abnormal intracellular protein transport and pigmentation
- Chediak-Higashi syndrome genes (LYST/CHS1) mutation
- Abnormal organelle trafficking and fusion
- Defective lysosome functions
- Neutrophils and macrophages with normal phagocytic function but delayed fusion of phagosomes with lysosomes
- NK cell and T cell cytotoxicity markedly decreased due to defective exocytosis of granules
- Melanosome defects
Clinical features
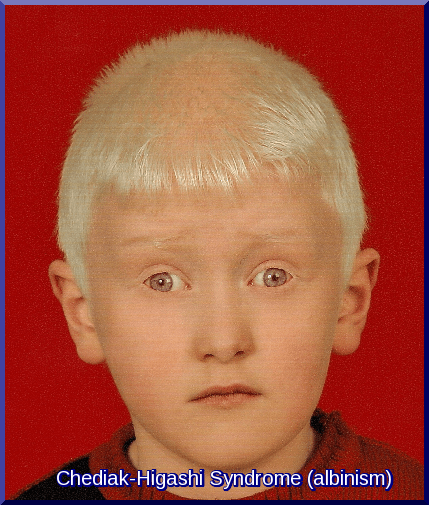
- Early presentations
- Nonpigmented skin, blonde hair, blue eyes (partial oculocutaneous albinism)
- Recurrent bacterial infections
- Coagulation defects, usually mild
- Adenopathy, aphthae, gingivitis, etc
- Progressive neurological dysfunction: peripheral neuropathy, weakness, ataxia, tremor, cranial nerve palsies, intellectual decline, etc
- Hemophagocytic lymphohistiocytosis/accelerated phase
{kind=link}
Key laboratory findings
- Neutropenia
- Markedly reduced or absent NK cytotoxicity
- Abnormal platelet aggregation
- Thrombocytopenia in accelerated phase
- Giant granules in leukocytes, fibroblasts, Schwann cells, muscle cells, melanocytes
{kind=link}
Genetic abnormality
- LYST/CHS1: Non-sense or null mutation
- No protein produced
Diagnosis
- History: early onset of recurrent bacterial infection, partial oculocutaneous albinism
- Lab: Giant granules in leukocytes
- Genetic testing for LYST/CHS1
Managements
- Hematopoietic cell transplantation
- Other supportive treatments: antibiotics, granulocyte colony stimulating factor (G-CSF), etc
- Treat Hemophagocytic lymphohistiocytosis/accelerated phase
Back to primary immunodeficiency disorders
Back to contents
Comments
Post a Comment