Epidermolysis bullosa
Epidermolysis bullosa
Updated: 02/09/2021
© Jun Wang, MD, PhD
General features
- Group of inherited disorders
- Onset at birth or early infancy
- Classified based on level of tissue separation
- Age of onset, possible inciting factor and family history of blistering for diagnosis
- NOT caused by inflammation
Pathogenesis
- Defects in cytokeratin filaments network
- Fragile basal keratinocytes, prompt to trauma-induced rupture
Epidermolysis
bullosa simplex
- Most common type
- Autosomal dominant mutations in keratin 14 or 5
- Intraepidermal bullae with basal cell degeneration
{kind=link}
Epidermolysis
bullosa junctional type
- Autosomal recessive
- Defects of laminin 5
- Blisters within lamina lucida
- Skin may appear normal
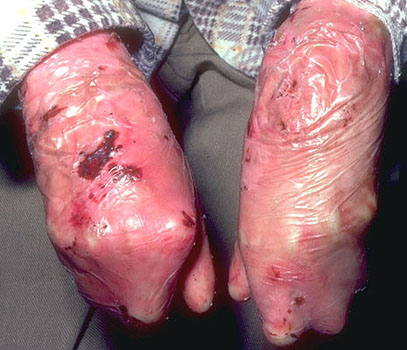
Epidermolysis
bullosa dystrophic type
- Autosomal dominant or recessive
- Defects in collagen VII, dermal side anchoring fibril protein
- Subepidermal blisters beneath lamina densa
- Digital fusion “mitten deformity”
{kind=link}
{kind=link}
Kindler
syndrome
- Autosomal recessive
- Congenital acral skin blistering, photosensitivity, progressive poikiloderma and diffuse cutaneous atrophy
- Loss-of-function mutation of FERMT1
- Defect in actin-ECM linkage
- Blisters at multiple levels (intra-lamina lucida and sub-lamina densa)
Genetic defects
- Simplex: keratin 5 and 14
- Junctional: laminin 5
- Dystrophic: collagen VII
- Kindler syndrome: actin-ECM linkage
Management
- Preventive and supportive management
Back to contents
Comments
Post a Comment